Peter G. Mayberry05.04.17
As required under provisions of the 21st Century Cures Act which was enacted late last year (Public Law 114-255), the U.S. Food and Drug Administration (FDA) has published lists of Class I and Class II medical devices to be exempted from premarket notification requirements under the agency’s 510(k) approval process. Both lists contain devices made from nonwovens or containing nonwovens.
Premarket approval is typically a costly and time-consuming process and, according to FDA, exemptions for Class I and Class II are intended to reduce regulatory burdens and expense borne by manufacturers for devices where the safety and effectiveness of the device is well established.
For members of the nonwovens industry who manufacture medical devices—especially drapes/gowns, pads, etc.—the ultimate result of this FDA action ought to be welcome, but FDA is also taking comments on whether the list of Class II devices should be “modified.“ This could serve as an opportunity for manufacturers of Class II devices that are not included on the published list to seek addition(s). Otherwise, PL 114-255 requires that similar lists be drawn up again no later than 2021 which will present another opportunity.
At Issue
Medical devices allowed for sale in the U.S. must be classified by FDA into one of three regulatory categories: Class I, Class II or Class III. FDA classification of specific devices is determined by the amount of regulation necessary to provide a “reasonable assurance of the device’s safety and effectiveness.” Currently, the safety and effectiveness of Class I devices is usually well known and can be sustained through a system of “General Controls.”
Devices are placed into Class II—also known as “Special Controls” – if General Controls, alone, can’t provide reasonable safety/effectiveness assurances but there is sufficient information to establish that special controls will provide these assurances. Class III devices cannot meet Class I or Class II standards but, otherwise, meet life-sustaining, life-supporting or similar other criteria. Virtually all medical devices made from or containing nonwovens are categorized as Class I or Class II.
One major difference between Class I and Class II is devices categorized as Class II have to undergo costly and time-consuming premarket approval requirements when they are modified, updated or altered—whereas manufacturers only need to notify FDA when similar changes are made to Class I devices.
According to FDA,, exemption from 510(k) requirements for Class I and Class II devices means manufacturers of those devices “will no longer have to invest time and resources in 510(k) submissions…including preparation of documents and data for submission to FDA, payment of user fees associated with 510(k) submissions and responding to questions and requests for additional information from FDA during 510(k) review.”
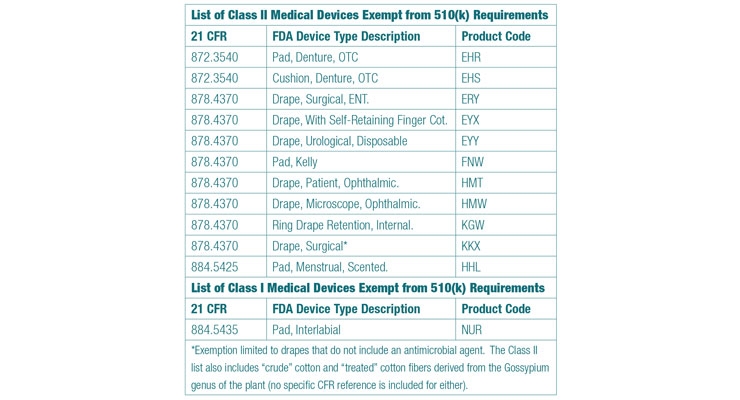
The adjoining table is gleaned from reviewing larger lists of Class I and Class II devices that FDA intends to exempt from 510(k) requirements. Interested readers are encouraged to review both lists to ensure comprehensiveness however. The Class II list is published in the March 14, 2017, addition of the Federal Register, and the Class I list can be found in the April 13 edition.
It’s noteworthy that the Class I list was published as a “Notice” by FDA whereas the Class II list was published as a “Final Rule.” The difference being FDA doesn’t indicate if it will accept public comment on the Class I list, but is encouraging comment on the list for Class II.
For Class II devices, a number of factors FDA may determine whether a 510(k) is necessary. Factors of “safety” and “effectiveness” are discussed in a 1998 FDA guidance document entitled “Procedures for Class II Device Exemptions from Premarket Notification: Guidance for Industry and CDRH Staff.’’
FDA stresses that an exemption from premarket notification requirements for a Class II device does not make the device exempt from any other statutory or regulatory requirements (unless such exemption is explicitly provided by order or regulation). Moreover, FDA’s initial determination that premarket notification isn’t needed to provide reasonable assurance of safety and effectiveness for Class II devices is based, in part, on the assurance of safety and effectiveness that accompany current good manufacturing practice requirements and other standards.
FDA will accept comment on the Class II list until midnight, Eastern, on May 15, 2017. Comments received by mail/hand delivery/courier must be postmarked—or have a delivery service acceptance receipt—on or before May 15. All comments should reference FDA docket number FDA-2017-N-1129. Comments submitted electronically should be uploaded to regulations.gov.
Type the FDA docket number into the search engine and follow the instructions for submitting comments.Comments submitted electronically, including attachments, will be posted to the docket as submitted, and warns not to include personal or business-sensitive information in a comment.
Partly due to its failure in reporting timely test performance results to the U.S. Food and Drug Administration (FDA), Halyard Health, Inc. – a 2014 spin off from Kimberly-Clark’s health care business – has been found liable by a California court for $454 million in damages related to impermeability claims the company made in marketing its MicroCool line of surgical gowns. Issues underlying this lawsuit were featured during an episode of the CBS news program “60 Minutes” which took the company to task for not informing FDA, and others, about test results indicating strikethrough incidents and other deficiencies during company-sponsored laboratory evaluations.
Premarket approval is typically a costly and time-consuming process and, according to FDA, exemptions for Class I and Class II are intended to reduce regulatory burdens and expense borne by manufacturers for devices where the safety and effectiveness of the device is well established.
For members of the nonwovens industry who manufacture medical devices—especially drapes/gowns, pads, etc.—the ultimate result of this FDA action ought to be welcome, but FDA is also taking comments on whether the list of Class II devices should be “modified.“ This could serve as an opportunity for manufacturers of Class II devices that are not included on the published list to seek addition(s). Otherwise, PL 114-255 requires that similar lists be drawn up again no later than 2021 which will present another opportunity.
At Issue
Medical devices allowed for sale in the U.S. must be classified by FDA into one of three regulatory categories: Class I, Class II or Class III. FDA classification of specific devices is determined by the amount of regulation necessary to provide a “reasonable assurance of the device’s safety and effectiveness.” Currently, the safety and effectiveness of Class I devices is usually well known and can be sustained through a system of “General Controls.”
Devices are placed into Class II—also known as “Special Controls” – if General Controls, alone, can’t provide reasonable safety/effectiveness assurances but there is sufficient information to establish that special controls will provide these assurances. Class III devices cannot meet Class I or Class II standards but, otherwise, meet life-sustaining, life-supporting or similar other criteria. Virtually all medical devices made from or containing nonwovens are categorized as Class I or Class II.
One major difference between Class I and Class II is devices categorized as Class II have to undergo costly and time-consuming premarket approval requirements when they are modified, updated or altered—whereas manufacturers only need to notify FDA when similar changes are made to Class I devices.
According to FDA,, exemption from 510(k) requirements for Class I and Class II devices means manufacturers of those devices “will no longer have to invest time and resources in 510(k) submissions…including preparation of documents and data for submission to FDA, payment of user fees associated with 510(k) submissions and responding to questions and requests for additional information from FDA during 510(k) review.”
The adjoining table is gleaned from reviewing larger lists of Class I and Class II devices that FDA intends to exempt from 510(k) requirements. Interested readers are encouraged to review both lists to ensure comprehensiveness however. The Class II list is published in the March 14, 2017, addition of the Federal Register, and the Class I list can be found in the April 13 edition.
It’s noteworthy that the Class I list was published as a “Notice” by FDA whereas the Class II list was published as a “Final Rule.” The difference being FDA doesn’t indicate if it will accept public comment on the Class I list, but is encouraging comment on the list for Class II.
For Class II devices, a number of factors FDA may determine whether a 510(k) is necessary. Factors of “safety” and “effectiveness” are discussed in a 1998 FDA guidance document entitled “Procedures for Class II Device Exemptions from Premarket Notification: Guidance for Industry and CDRH Staff.’’
FDA stresses that an exemption from premarket notification requirements for a Class II device does not make the device exempt from any other statutory or regulatory requirements (unless such exemption is explicitly provided by order or regulation). Moreover, FDA’s initial determination that premarket notification isn’t needed to provide reasonable assurance of safety and effectiveness for Class II devices is based, in part, on the assurance of safety and effectiveness that accompany current good manufacturing practice requirements and other standards.
FDA will accept comment on the Class II list until midnight, Eastern, on May 15, 2017. Comments received by mail/hand delivery/courier must be postmarked—or have a delivery service acceptance receipt—on or before May 15. All comments should reference FDA docket number FDA-2017-N-1129. Comments submitted electronically should be uploaded to regulations.gov.
Type the FDA docket number into the search engine and follow the instructions for submitting comments.Comments submitted electronically, including attachments, will be posted to the docket as submitted, and warns not to include personal or business-sensitive information in a comment.
Partly due to its failure in reporting timely test performance results to the U.S. Food and Drug Administration (FDA), Halyard Health, Inc. – a 2014 spin off from Kimberly-Clark’s health care business – has been found liable by a California court for $454 million in damages related to impermeability claims the company made in marketing its MicroCool line of surgical gowns. Issues underlying this lawsuit were featured during an episode of the CBS news program “60 Minutes” which took the company to task for not informing FDA, and others, about test results indicating strikethrough incidents and other deficiencies during company-sponsored laboratory evaluations.









